- Implement & Learn
Implementation Products - Resources
- For Consultants
- About Us
EU MDR Expert - Contact Us

Updated: October 19, 2023.
EU MDR stands for European Union Medical Device Regulation. It is a set of regulations that govern the safety and performance requirements of medical devices in the European market. Besides “What does MDR stand for?”, many other questions need to be answered to show you the big picture of the EU Medical Device Regulation. What is the purpose of the MDR? Who needs to comply with the EU MDR? How many EU countries are impacted by the MDR? Where is the EU MDR applicable? Learn more in this article.
The aim of the EU MDR is to ensure the highest level of patient safety and provide greater transparency and traceability of medical devices throughout their lifecycles. The EU MDR was released by the European Parliament and the Council of the European Union. The intent of the European medical device regulations is to ensure a high standard of safety and quality for medical devices that are produced in, or supplied to, member countries of the European Union.
This regulatory framework is intended to better identify medical devices, as well as standardizing data and technological advances through an EU database (EUDAMED). The EU MDR is intended to be a regulatory framework for medical devices that can sustainably ensure health & safety while still encouraging innovation.
The EU MDR does not incorporate In Vitro Diagnostic Regulation (IVDR). For a detailed comparison of these two regulations, read the article Comparison of the EU MDR and IVDR regulations.
The EU MDR comes with several benefits for both patients and manufacturers. Firstly, it ensures a higher level of patient safety by enforcing stricter regulations on medical devices. Secondly, it promotes greater transparency and traceability of medical devices throughout their lifecycles, which helps in identifying and addressing any potential safety issues or recalls. Thirdly, it enhances the overall quality and reliability of medical devices available in the European market.
Additionally, the EU MDR encourages innovation by promoting the development of safer and more effective medical devices. Lastly, it harmonizes the regulations across the European Union, making it easier for manufacturers to navigate the market.
MDD stands for Medical Device Directive (93/47/EEC), which was the previous regulatory framework for medical devices in the European Union. The Medical Device Directive (MDD) outlined the safety and performance requirements for medical devices sold in the EU market. The MDD had been in place for almost 25 years before it was replaced by the new European Union Medical Device Regulation (MDR), issued in 2017.
MDR 2017/745, on the other hand, stands for Medical Device Regulation. It is the current regulatory framework that replaced the MDD. The MDR introduces stricter regulations and requirements to ensure the safety, quality, and reliability of medical devices in the European market.
The EU MDR was published on May 5, 2017, and there was a three-year transition period for companies to comply with the new MDR requirements, until May 2020. Then, due to the COVID situation, this date was extended to May 2021.
For products that have not been on the EU market under the Medical Device Directive (MDD), the May 2021 date for the EU MDR means that they will need to comply with the new regulation before they can be placed on the market. This includes undergoing a conformity assessment process and obtaining a valid CE mark.

However, due to various challenges faced by stakeholders, such as the complexity of implementing the new regulations and the limited availability of notified bodies, the European Union decided to postpone its application.
The application of the MDR for the devices that are already on the market under the MDD was extended in March 2023 to the end of 2027 or 2028 (depending on the medical device classification), to allow manufacturers, notified bodies, and other parties involved to adequately prepare for the new requirements. This extension provides additional time for manufacturers to ensure compliance with the stricter regulations and to complete the necessary conformity assessments for their medical devices.
The extension of the MDR aims to minimize disruptions in the availability of medical devices in the European market and to ensure a smooth transition from the previous regulatory framework (MDD) to the new one. It also allows a more phased and controlled implementation process, ensuring that all stakeholders are well prepared and can meet the new obligations outlined in the MDR.
Overall, the extension of the MDR provides a realistic timeline for all parties involved to adapt to the new regulatory landscape and to continue providing safe and high-quality medical devices to patients in the European Union.
Under the EU MDR, medical device manufacturers are required to demonstrate compliance with certain essential requirements and undergo a conformity assessment process. This involves conducting clinical evaluations, risk assessments, and post-market surveillance activities to ensure ongoing compliance and safety of the devices.
Additionally, the EU MDR introduces new classifications for medical devices based on their risk level, with stricter requirements for higher-risk devices. It also places greater emphasis on traceability and transparency in the supply chain, with the introduction of unique device identifiers (UDIs) and the European database on medical devices (EUDAMED).
Non-compliance with the EU MDR can have serious consequences for medical device manufacturers, including the inability to sell their products in the EU market, financial penalties, and damage to their reputation. Therefore, it is crucial for manufacturers to understand and adhere to the requirements of EU MDR compliance to ensure the safety and effectiveness of their medical devices.
Overall, EU MDR compliance plays a vital role in ensuring the quality and safety of medical devices in the European Union, ultimately protecting the health and well-being of patients.
The MDR certification is required for medical device manufacturers to legally market and sell their products in the EU. To obtain MDR certification, manufacturers must comply with strict regulatory requirements and undergo a thorough assessment of their devices. This includes conducting clinical evaluations, implementing a Quality Management System, and meeting specific labeling and documentation requirements.
Many companies use ISO 13485 as a way to implement this QMS, as this is the only QMS standard on the EU harmonized lists; therefore, it is the best way to implement the QMS as it relates to the EU MDR. The purpose of MDR certification is to enhance patient safety and improve the overall quality of medical devices available in the EU market. It ensures that medical devices are designed, manufactured, and used in a way that minimizes risks and maximizes benefits for patients.
Having MDR certification demonstrates that a medical device meets the necessary safety and performance standards set by the EU regulatory authorities. It provides reassurance to healthcare professionals and patients that the device has undergone rigorous testing and evaluation. Overall, MDR certification plays a crucial role in ensuring the safety and effectiveness of medical devices in the EU, promoting public health and maintaining confidence in the healthcare system.
MDR technical documentation refers to the set of documents that provide comprehensive information about a medical device and its design, manufacturing, and performance characteristics, stated in Annex II and Annex III. This documentation plays a crucial role in ensuring compliance with regulatory requirements and demonstrating the safety and effectiveness of the medical device.
The MDR technical documentation typically includes:
It is important to note that the MDR technical documentation requirements are stringent and vary depending on the classification of the medical device. Manufacturers must ensure that the documentation is comprehensive, up to date, and readily available for review by regulatory authorities.
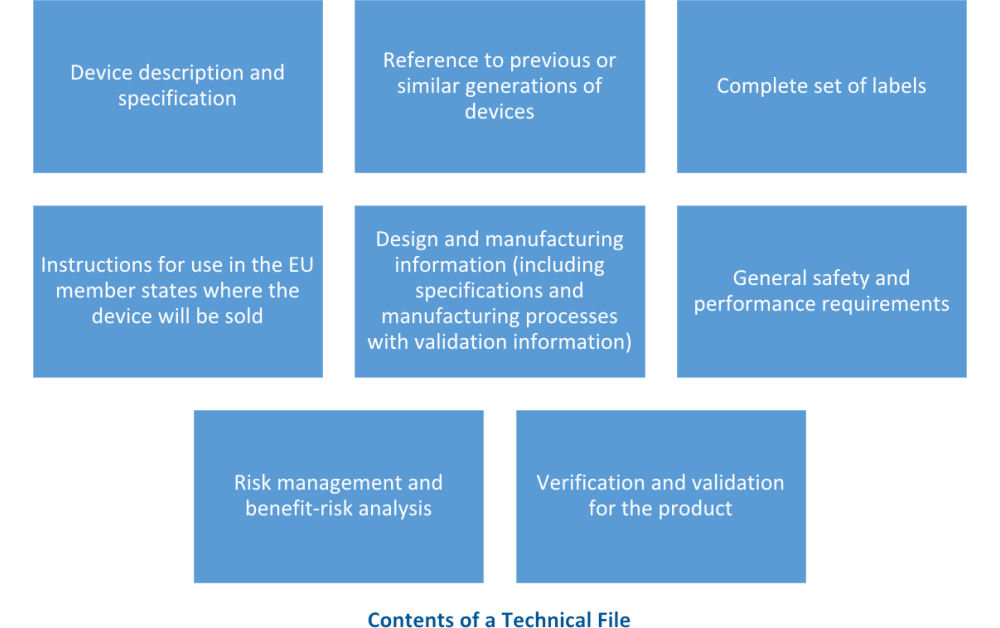
This practice matches with ISO 13485:2016 clause 4.2.3, which requires device manufacturers to create a technical file, or a medical device file.
The relationship between the MDR and healthcare is crucial, as it sets the standards and regulations that medical device manufacturers must follow to ensure the safety and effectiveness of their products. Healthcare professionals rely on medical devices to diagnose, treat, and monitor patients, and the MDR helps to ensure that these devices meet the necessary quality and safety standards.
By enforcing strict regulations and requirements, the MDR aims to enhance patient safety and improve the overall quality of healthcare by ensuring that medical devices are safe, effective, and perform as intended. Compliance with the MDR is essential for medical device manufacturers to gain market access within the European Union and to maintain the trust of healthcare providers and patients.
Overall, the European medical device regulations play a crucial role in safeguarding the relationship between healthcare and medical devices, ensuring that healthcare professionals have access to safe and reliable medical devices to deliver high-quality care to patients.






 Mark Hammar
Mark Hammar 